hide abstracts :: all publications on PubMed

PLoS Computational Biology 5(2):e1000290
Dudman JT and Nolan MF
Stochastically gating ion channels enable patterned spike firing through activity-dependent modulation of spike probability.
Many computations in neural circuits rely upon appropriate patterning of action potentials. The patterns of action potentials a neuron fires are shaped by its complement of membrane ion channels. However, while the total ionic current across a neuron’s membrane is determined by stochastic transitions between open and closed states of many individual ions channels, most neuronal models are deterministic and consider only the average behavior of populations of ion channels. We show that a model of stellate neurons found in layer II of the entorhinal cortex, when implemented using stochastically gating ion channels, accounts well for experimentally described properties of these neurons, including firing of clustered patterns of action potentials and functional changes caused by deletion of HCN1 channels. Analysis of the model shows that clustered patterns of action potentials arise through transient increases in the probability of spike initiation during a brief time window following recovery from a preceding action potential. This model provides an example of a general mechanism for patterning of neuronal activity through brief activity-dependent changes in spike probability and may help explain the patterns of spikes fired by entorhinal neurons that encode spatial location in behaving animals.
Neuron 56(6):1076-1089
Tsay D, Dudman JT, and Siegelbaum SA
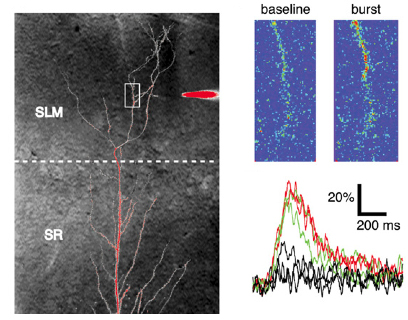
HCN1 constrains synaptically-evoked calcium events in
distal dendrites of CA1 pyramidal neurons.
Distal dendrites of hippocampal CA1 pyramidal neurons receive direct sensory information
that is relevant to spatial learning and memory through the perforant path synapses from
the entorhinal cortex. The HCN1 hyperpolarization-activated cation channels are targeted
to these distal dendrites where they act as an inhibitory constraint of synaptic
integration and long-term plasticity. However, since HCN channels contribute a
depolarizing current, the mechanism of their inhibitory action remains unclear. Here we
report that HCN1 constrains synaptically-evoked distal dendritic Ca2+ spikes, which have
previously been implicated in the induction of LTP at the perforant path synapses.
Experimental and computational results indicate that resting HCN channels provide a
steady-state depolarization that increases resting inactivation of T-type and N-type
voltage-gated Ca2+ channels, thereby limiting distal Ca2+ influx. This mechanism by
which Ih constrains voltage-gated Ca2+ channel availability may represent a general
means by which these channels regulate dendritic excitability and calcium electrogenesis
at distal compartments of pyramidal neurons where high densities of h-channels are found.

Neuron 56(5):866-79
Dudman JT, Tsay D, and Siegelbaum SA
A novel role for distal synaptic inputs: instructive
signals for hippocampal synaptic plasticity.
Synaptic potentials originating at distal dendritic locations are severely attenuated
when they reach the soma and, thus, are poor at driving somatic spikes. Nonetheless,
such inputs often convey essential information, suggesting that distal inputs may be
important for compartmentalized dendritic signaling. Here we report a new plasticity
rule in which stimulation of the distal perforant path inputs to hippocampal CA1
pyramidal neurons induces long-term potentiation at proximal Schaffer collateral
synapses when the two inputs are paired at a precise interval. This subthreshold form
of heterosynaptic plasticity occurs in the absence of somatic spiking but requires
activation of both NMDA receptors and release of Ca2+ from internal stores. Our results
suggest that direct sensory information arriving at distal CA1 synapses through the
perforant path may serve a novel function: providing compartmentalized, instructive
signals that assess the saliency of mnemonic information propagated through the
hippocampal circuit to proximal synapses.

Journal of Neuroscience 27(46):12440-51
Nolan MF, Dudman JT, Dodson, PD and Santoro B
HCN1 channels control resting and active integrative
properties of stellate cells from layer II of the entorhinal cortex.
Whereas recent studies have elucidated principles for representation of information
within the entorhinal cortex, less is known about the molecular basis for information
processing by entorhinal neurons. The HCN1 gene encodes ion channels that mediate
hyperpolarization-activated currents (Ih) that control synaptic integration and influence
several forms of learning and memory. We asked if HCN1 channels may control processing
of information by stellate shaped cells found within layer II of the entorhinal cortex.
Axonal projections from this non-pyramdial class of principle neuron form a major
component of the synaptic input to the dentate gyrus of the hippocampus. To investigate
the influence of HCN1 channels on resting and active properties of stellate neurons we
carried out whole-cell recordings in horizontal brain slices prepared from adult
wild-type and HCN1 knockout mice. We find that HCN1 channels are required for rapid
and full activation of hyperpolarization-activated currents in stellate neurons. HCN1
channels dominate the membrane conductance at rest and suppress low frequency (< 4 Hz)
components of spontaneous and evoked membrane potential activity. In addition, we find
that when stellate cells receive continuous depolarizing input sufficient for firing of
repeated action potentials, HCN1 channels control the pattern of spike output by
promoting recovery of the spike afterhyperpolarization. These data suggest that HCN1
channels may control the input to the hippocampal dentate gyrus by distinct actions on
integration by entorhinal stellate cells during resting and active states.
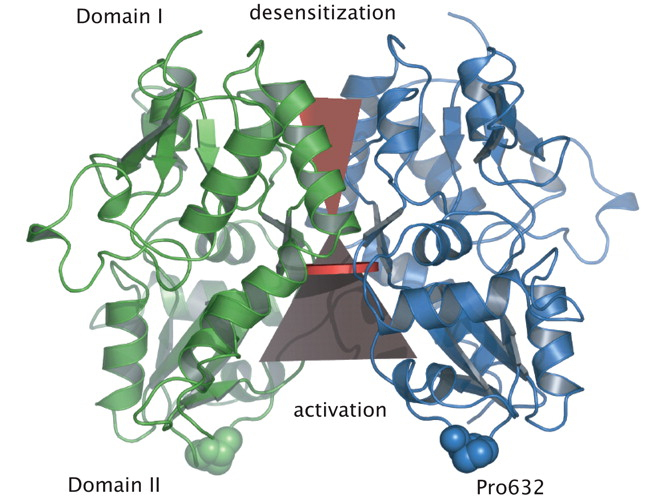
Journal of Neuroscience 25(39):9027-36
Jin R, Clark S, Weeks AM, Dudman JT, Gouaux E, Partin KM
Mechanism of positive allosteric modulators acting
on AMPA receptors
Ligand-gated ion channels involved in the modulation of synaptic strength are the AMPA,
kainate, and NMDA glutamate receptors. Small molecules that potentiate AMPA receptor
currents relieve cognitive deficits caused by neurodegenerative diseases such as
Alzheimer's disease and show promise in the treatment of depression. Previously, there
has been limited understanding of the molecular mechanism of action for AMPA receptor
potentiators. Here we present cocrystal structures of the glutamate receptor GluR2 S1S2
ligand-binding domain in complex with aniracetam [1-(4-methoxybenzoyl)-2-pyrrolidinone]
or CX614 (pyrrolidino-1,3-oxazino benzo-1,4-dioxan-10-one), two AMPA receptor potentiators
that preferentially slow AMPA receptor deactivation. Both potentiators bind within the
dimer interface of the nondesensitized receptor at a common site located on the twofold
axis of molecular symmetry. Importantly, the potentiator binding site is adjacent to the
"hinge" in the ligand-binding core "clamshell" that undergoes conformational rearrangement
after glutamate binding. Using rapid solution exchange, patch-clamp electrophysiology
experiments, we show that point mutations of residues that interact with potentiators in
the cocrystal disrupt potentiator function. We suggest that the potentiators slow
deactivation by stabilizing the clamshell in its closed-cleft, glutamate-bound conformation.

Biological Psychiatry 57(9):1041-1051
MacDonald M, Eaton ME, Dudman JT, Konradi C
Antipsychotic drugs elevate mRNA levels of presynaptic proteins in the frontal
cortex of the rat.
Molecular adaptations are believed to contribute to the mechanism of action of
antipsychotic drugs (APDs). We attempted to establish common gene regulation patterns
induced by chronic treatment with APDs. METHODS: Gene expression analysis was performed
with the Affymetrix U34A array in the frontal cortex (FC) and the striatum of rats
chronically treated with two concentrations of either clozapine or haloperidol. Key data
were verified with real-time quantitative polymerase chain reaction. RESULTS: Many genes
in the FC affected by APD-treatment contribute to similar functions. mRNAs coding for
synaptic vesicle docking- and microtubule-associated proteins were upregulated; mRNAs for
serine-threonine protein phosphatases were downregulated, whereas the serine-threonine
kinases protein kinase A, protein kinase C, and calcium/calmodulin kinase II alpha and
IV were upregulated, indicating increased potential for protein phosphorylation. In the
striatum, altered gene expression was less focused on genes of particular function or
location, and the high concentration of haloperidol had a different gene expression
profile than any of the other APD treatments. CONCLUSION: We found an increase in the
transcription of genes coding for proteins involved in synaptic plasticity and synaptic
activity in the FC. We furthermore found that the gene expression profile of APDs is
different between FC and striatum.
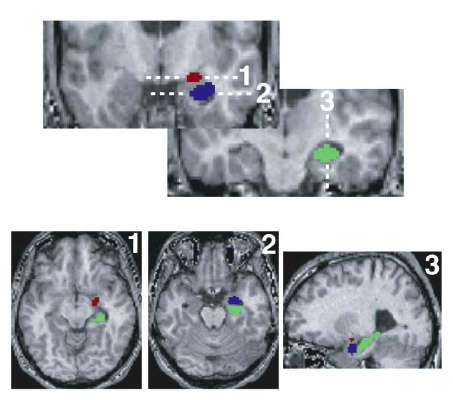
Neuron 44(6):1043-1055
Etkin A, Klemenhagen KC,
Dudman JT, Rogan MT, Hen R, Kandel ER, Hirsch J
Individual Differences in Trait Anxiety Predict the Response of the Basolateral
Amygdala to Unconsciously Processed Fearful Faces
Responses to threat-related stimuli are influenced by conscious and unconscious processes,
but the neural systems underlying these processes and their relationship to anxiety have
not been clearly delineated. Using fMRI, we investigated the neural responses associated
with the conscious and unconscious (backwardly masked) perception of fearful faces in
healthy volunteers who varied in threat sensitivity (Spielberger trait anxiety scale).
Unconscious processing modulated activity only in the basolateral subregion of the
amygdala, while conscious processing modulated activity only in the dorsal amygdala
(containing the central nucleus). Whereas activation of the dorsal amygdala by conscious
stimuli was consistent across subjects and independent of trait anxiety, activity in the
basolateral amygdala to unconscious stimuli, and subjects' reaction times, were predicted
by individual differences in trait anxiety. These findings provide a biological basis for
the unconscious emotional vigilance characteristic of anxiety and a means for
investigating the mechanisms and efficacy of treatments for anxiety.

Cell 119(5):719-32
Nolan MF, Malleret G,
Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G,
Siegelbaum SA, Kandel ER, Morozov A
A behaivoral
role for dendritic integration: HCN1 channels constrain spatial memory
and plasticity at inputs to distal dendrites of CA1 pyramidal neurons
The importance of long-term synaptic plasticity as a cellular substrate for learning and
memory is well established. By contrast, little is known about how learning and memory are
regulated by voltage-gated ion channels that integrate synaptic information. We investigated
this question using mice with general or forebrain-restricted knockout of the HCN1 gene,
which we find encodes a major component of the hyperpolarization-activated inward current
(Ih) and is an important determinant of dendritic integration in hippocampal CA1 pyramidal
cells. Deletion of HCN1 from forebrain neurons enhances hippocampal-dependent learning and
memory, augments the power of theta oscillations, and enhances long-term potentiation (LTP)
at the direct perforant path input to the distal dendrites of CA1 pyramidal neurons, but
has little effect on LTP at the more proximal Schaffer collateral inputs. We suggest that
HCN1 channels constrain learning and memory by regulating dendritic integration of distal
synaptic inputs to pyramidal cells.

Cell 115(5):551-64
Nolan MF, Malleret G, Lee KH,
Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER,
Morozov A
The hyperpolarization-activated HCN1 channels is
important for motor learning and neuronal integration by cerebellar Purkinje cells
In contrast to our increasingly detailed understanding of how synaptic plasticity provides
a cellular substrate for learning and memory, it is less clear how a neuron's voltage-gated
ion channels interact with plastic changes in synaptic strength to influence behavior. We
find, using generalized and regional knockout mice, that deletion of the HCN1 channel
causes profound motor learning and memory deficits in swimming and rotarod tasks. In
cerebellar Purkinje cells, which are a key component of the cerebellar circuit for learning
of correctly timed movements, HCN1 mediates an inward current that stabilizes the
integrative properties of Purkinje cells and ensures that their input-output function is]
independent of the previous history of their activity. We suggest that this nonsynaptic
integrative function of HCN1 is required for accurate decoding of input patterns and
thereby enables synaptic plasticity to appropriately influence the performance of motor
activity.
Journal of Neurochemistry 87(4):922-34
Dudman JT, Eaton ME, Rajadhyaksha A, Macias W, Taher M, Barczak A,
Kameyama K, Huganir R, Konradi C
Dopamine D1 receptors
mediate CREB phosphorylation via phosphorylation of NMDA receptor at Ser897-NR1
Addictive drugs such as amphetamine and cocaine stimulate the dopaminergic system, activate
dopamine receptors and induce gene expression throughout the striatum. The signal
transduction pathway leading from dopamine receptor stimulation at the synapse to gene
expression in the nucleus has not been fully elucidated. Here, we present evidence that D1
receptor stimulation leads to phosphorylation of the transcription factor Ca2+ and cyclic
AMP response element binding protein (CREB) in the nucleus by means of NMDA
receptor-mediated Ca2+ signaling. Stimulation of D1 receptors induces the phosphorylation
of Ser897 on the NR1 subunit by protein kinase A (PKA). This phosphorylation event is
crucial for D1 receptor-mediated CREB phosphorylation. Dopamine cannot induce CRE-mediated
gene expression in neurons transfected with a phosphorylation-deficient NR1 construct.
Moreover, stimulation of D1 receptors or increase in cyclic AMP levels leads to an increase
in cytosolic Ca2+ in the presence of glutamate, but not in the absence of glutamate,
indicating the ability of dopamine and cyclic AMP to facilitate NMDA channel activity. The
recruitment of the NMDA receptor signal transduction pathway by D1 receptors may provide a
general mechanism for gene regulation that is fundamental for mechanisms of drug addiction
and long-term memory.